
Preparing a Structure for Molecular Simulation
Prerequisites
Prior to starting this tutorial, make sure you have installed and are familiar with the following:
Tutorial
Background
It is paramount that protein structures (.pdb) are prepared properly prior to conducting molecular simulations. If the quality of the sturcture is poor, any analysis from the simulation data will likely be useless.
The most well-known database for the three-dimensional structural data of proteins is the Protein Data Bank. This data is typically obtained by experimental techniques such as X-ray crystallography and cryo-electron microscopy. These techniques are not perfect and consequently there will be residues in the protein for which coordinate data is missing.
This tutorial will walk through how we can use Modeller to generate a homology model based on multiple templates and introduce mutations into the model.
Introduction
Imagine that we are oncologists, and we are presented with a neuroblastoma patient who has a novel mutation, R1275Q, in the ALK gene. To investigate whether this mutation is the driver for this patient’s neuroblastoma, we would like to conduct a MD simulation. Namely, we want to conduct an MD simulation of the ALK kinase in the inactive conformation, with the R1275Q mutation, and analyze changes in the hydrogen bond network of the protein.
The first step is acquiring a structure of the inactive conformation of the ALK kinase (with no inhibitors bound). A quick google search shows us that the following is the entry of interest: 3LCS.
Multiple Template Homology Model
As expected, upon inspection of the PDB file, we find that there the following residues (of interest) are missing:
- 1084-1095
- 1400-1405
Modeller does provide native functionality for filling in these gaps, but an alternative strategy is looking for other ALK PDBs in the database where perhaps other regions of the protein are missing but coordinates for residues 1084-1095 and 1400-1405 are known. With a little more research, we find that 4FNW has these coordinates. Thus, we can use both 3LCS and 4FNW in creating the most accurate structure for the inactive confromation of ALK.
Before starting, make sure sure that you create a directory for this tutorial, and save both of the PDB files (4FNW,3LCS) to this directory. We will now follow the “Multiple Templates” section of Modeller’s advanced tutorial to generate a homology model of ALK using multiple templates. As you are following along, if you have any questions regarding the scripts used, please refer to that tutorial.
-
First, it is necessary to put the target ALK sequence into the PIR format readable by Modeller. This can be done by copying the FASTA sequence from the 4FNW PDB entry and formatting as shown below. Save the text below as “ALK.ali” in the same directory.
>P1;ALK sequence:ALK:::::::0.00: 0.00 RTSTIMTDYNPNYSFAGKTSSISDLKEVPRKNITLIRGLGHGAFGEVYEGQVSGMPNDPSPLQVAVKTLPEVCSEQDELD FLMEALIISKLNHQNIVRCIGVSLQSLPRFILLELMAGGDLKSFLRETRPRPSQPSSLAMLDLLHVARDIACGCQYLEEN HFIHRDIAARNCLLTCPGPGRVAKIGDFGMARDIYRASYYRKGGCAMLPVKWMPPEAFMEGIFTSKTDTWSFGVLLWEIF SLGYMPYPSKSNQEVLEFVTSGGRMDPPKNCPGPVYRIMTQCWQHQPEDRPNFAIILERIEYCTQDPDVINTALPIEYGP LVEEEEK* -
The next step is to align the sequences of our two templates (4FNW and 3LCS). The following script will read in all of the sequences from the template PDB files, and then locally align the sequences multiple times, to generate an initial rough alignment and then improve upon it using more information. This alignemnt is then written out in both PIR and PAP formats. Save the following code as “salign.py” in the same directory, and then run the python script.
# Illustrates the SALIGN multiple structure/sequence alignment from modeller import * log.verbose() env = Environ() env.io.atom_files_directory = ['.', '../atom_files/'] aln = Alignment(env) for (code, chain) in (('3lcs', 'A'), ('4fnw', 'A')): mdl = Model(env, file=code, model_segment=('FIRST:'+chain, 'LAST:'+chain)) aln.append_model(mdl, atom_files=code, align_codes=code+chain) for (weights, write_fit, whole) in (((1., 0., 0., 0., 1., 0.), False, True), ((1., 0.5, 1., 1., 1., 0.), False, True), ((1., 1., 1., 1., 1., 0.), True, False)): aln.salign(rms_cutoff=3.5, normalize_pp_scores=False, rr_file='$(LIB)/as1.sim.mat', overhang=30, gap_penalties_1d=(-450, -50), gap_penalties_3d=(0, 3), gap_gap_score=0, gap_residue_score=0, dendrogram_file='fm00495.tree', alignment_type='tree', # If 'progresive', the tree is not # computed and all structues will be # aligned sequentially to the first feature_weights=weights, # For a multiple sequence alignment only # the first feature needs to be non-zero improve_alignment=True, fit=True, write_fit=write_fit, write_whole_pdb=whole, output='ALIGNMENT QUALITY') aln.write(file='templates.pap', alignment_format='PAP') aln.write(file='templates.ali', alignment_format='PIR') aln.salign(rms_cutoff=1.0, normalize_pp_scores=False, rr_file='$(LIB)/as1.sim.mat', overhang=30, gap_penalties_1d=(-450, -50), gap_penalties_3d=(0, 3), gap_gap_score=0, gap_residue_score=0, dendrogram_file='1is3A.tree', alignment_type='progressive', feature_weights=[0]*6, improve_alignment=False, fit=False, write_fit=True, write_whole_pdb=False, output='QUALITY') -
Now, we need to align our target sequence to the template structures. For this, we use a pairwise alignment algorthm as we do not want to change the existing alignment between the templates. Save the following code as “align2d_mult.py” in the same directory, and then run the python script.
from modeller import * log.verbose() env = Environ() env.libs.topology.read(file='$(LIB)/top_heav.lib') # Read aligned structure(s): aln = Alignment(env) aln.append(file='templates.ali', align_codes='all') aln_block = len(aln) # Read aligned sequence(s): aln.append(file='ALK.ali', align_codes='ALK') # Structure sensitive variable gap penalty sequence-sequence alignment: aln.salign(output='', max_gap_length=20, gap_function=True, # to use structure-dependent gap penalty alignment_type='PAIRWISE', align_block=aln_block, feature_weights=(1., 0., 0., 0., 0., 0.), overhang=0, gap_penalties_1d=(-450, 0), gap_penalties_2d=(0.35, 1.2, 0.9, 1.2, 0.6, 8.6, 1.2, 0., 0.), similarity_flag=True) aln.write(file='ALK-mult.ali', alignment_format='PIR') aln.write(file='ALK-mult.pap', alignment_format='PAP') -
Next, we build the new model for the ALK target sequence based on the alignment against the multiple templates. The atomic coordinates for each residue will be filled in based on overlap between our target sequence and that of the known templates. For regions where there is no overlap between the template, Modeller will guess the coordinates to fill in remaining gaps. Save the following code as “model_mult.py” in the same directory, and then run the python script.
from modeller import * from modeller.automodel import * env = Environ() a = AutoModel(env, alnfile='ALK-mult.ali', knowns=('3lcsA','4fnwA'), sequence='ALK') a.starting_model = 1 a.ending_model = 1 a.make() -
The final structure should be saved as ‘ALK.B99990001.pdb’ and have a molecular PDF (molpdf) value of 9932.03711. molpdf is the standard Modeller scoring function and simply a sum of all restraints applied during homology modeling. In this case, we only outputted one model so this value is not of importance to us. Modeller is capable of providing multiple possible models, and in that case, the model with the lowest molpdf (less restraints imposed) would be the best model. As a quick check, you can use VMD to visualize the structure and compare it to our templates.
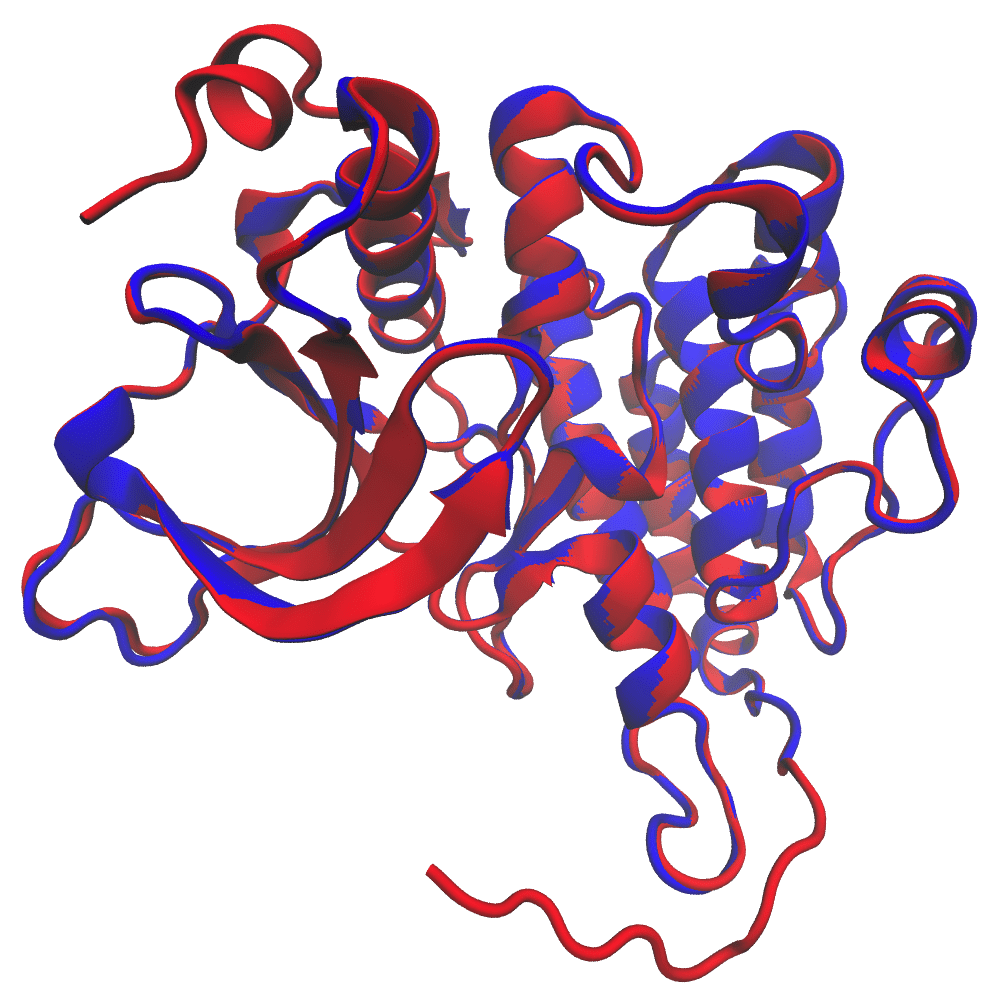
The structure generated by Modeller (color: red) overlaid with the 3LCS structure (color: blue)
And there you have it! We were able to successfully use Modeller to fill in the gaps of the 3LCS structure using the 4FNW template.
Mutate Model
Now that we have an accurate structure for the inactive conformation of ALK, the next step is to introduce the R1275Q mutation.
-
Prior to doing this, one thing you will notice is that Modeller renumbered our PDB from 1 whereas the original PDB starts from 1084. The following script will allow us to renumber our PDF prior to introducing the mutation of interest. Save the following code as “renumber_model.py” in the same directory, and then run the python script.
# Example for: model.rename_segments() # This will assign new PDB single-character chain id's to all the chains # in the input PDB file (here there are two 'chains': protein and the HETATM # water molecules). from modeller import * # Read the MODEL with all HETATM and water records (so there are two 'chains'): env = environ() env.io.atom_files_directory = ['../atom_files'] env.io.hetatm = True env.io.water = True #import .pdb file mdl = model(env, file='ALK.B99990001.pdb') # Assign new segment names and write out the new model. The residue numbers will only change for chain A which is our protein chain: mdl.rename_segments(segment_ids=('A', ' '),renumber_residues=[1084,1]) mdl.write(file='alk_renumbered.pdb') -
The outputted PDB should show up in the directory as “alk_renumbered.pdb”. The following script will next create a single in silico point mutation at the location indicated by the user and optimize the conformation of the mutant sidechain by conjugate gradient and MD simulation. Save the following code as “mutate_model.py” in the same directory.
import sys import os from modeller import * from modeller.optimizers import MolecularDynamics, ConjugateGradients from modeller.automodel import autosched # # mutate_model.py # # Usage: python mutate_model.py modelname respos resname chain > logfile # # Example: python mutate_model.py 1t29 1699 LEU A > 1t29.log # # # Creates a single in silico point mutation to sidechain type and at residue position # input by the user, in the structure whose file is modelname.pdb # The conformation of the mutant sidechain is optimized by conjugate gradient and # refined using some MD. # # Note: if the model has no chain identifier, specify "" for the chain argument. # def optimize(atmsel, sched): #conjugate gradient for step in sched: step.optimize(atmsel, max_iterations=200, min_atom_shift=0.001) #md refine(atmsel) cg = ConjugateGradients() cg.optimize(atmsel, max_iterations=200, min_atom_shift=0.001) #molecular dynamics def refine(atmsel): # at T=1000, max_atom_shift for 4fs is cca 0.15 A. md = MolecularDynamics(cap_atom_shift=0.39, md_time_step=4.0, md_return='FINAL') init_vel = True for (its, equil, temps) in ((200, 20, (150.0, 250.0, 400.0, 700.0, 1000.0)), (200, 600, (1000.0, 800.0, 600.0, 500.0, 400.0, 300.0))): for temp in temps: md.optimize(atmsel, init_velocities=init_vel, temperature=temp, max_iterations=its, equilibrate=equil) init_vel = False #use homologs and dihedral library for dihedral angle restraints def make_restraints(mdl1, aln): rsr = mdl1.restraints rsr.clear() s = Selection(mdl1) for typ in ('stereo', 'phi-psi_binormal'): rsr.make(s, restraint_type=typ, aln=aln, spline_on_site=True) for typ in ('omega', 'chi1', 'chi2', 'chi3', 'chi4'): rsr.make(s, restraint_type=typ+'_dihedral', spline_range=4.0, spline_dx=0.3, spline_min_points = 5, aln=aln, spline_on_site=True) #first argument modelname, respos, restyp, chain, = sys.argv[1:] log.verbose() # Set a different value for rand_seed to get a different final model env = Environ(rand_seed=-49837) env.io.hetatm = True #soft sphere potential env.edat.dynamic_sphere=False #lennard-jones potential (more accurate) env.edat.dynamic_lennard=True env.edat.contact_shell = 4.0 env.edat.update_dynamic = 0.39 # Read customized topology file with phosphoserines (or standard one) env.libs.topology.read(file='$(LIB)/top_heav.lib') # Read customized CHARMM parameter library with phosphoserines (or standard one) env.libs.parameters.read(file='$(LIB)/par.lib') # Read the original PDB file and copy its sequence to the alignment array: mdl1 = Model(env, file=modelname) ali = Alignment(env) ali.append_model(mdl1, atom_files=modelname, align_codes=modelname) #set up the mutate residue selection segment s = Selection(mdl1.chains[chain].residues[respos]) #perform the mutate residue operation s.mutate(residue_type=restyp) #get two copies of the sequence. A modeller trick to get things set up ali.append_model(mdl1, align_codes=modelname) # Generate molecular topology for mutant mdl1.clear_topology() mdl1.generate_topology(ali[-1]) # Transfer all the coordinates you can from the template native structure # to the mutant (this works even if the order of atoms in the native PDB # file is not standard): #here we are generating the model by reading the template coordinates mdl1.transfer_xyz(ali) # Build the remaining unknown coordinates mdl1.build(initialize_xyz=False, build_method='INTERNAL_COORDINATES') #yes model2 is the same file as model1. It's a modeller trick. mdl2 = Model(env, file=modelname) #required to do a transfer_res_numb #ali.append_model(mdl2, atom_files=modelname, align_codes=modelname) #transfers from "model 2" to "model 1" mdl1.res_num_from(mdl2,ali) #It is usually necessary to write the mutated sequence out and read it in #before proceeding, because not all sequence related information about MODEL #is changed by this command (e.g., internal coordinates, charges, and atom #types and radii are not updated). mdl1.write(file=modelname+restyp+respos+'.tmp') mdl1.read(file=modelname+restyp+respos+'.tmp') #set up restraints before computing energy #we do this a second time because the model has been written out and read in, #clearing the previously set restraints make_restraints(mdl1, ali) #a non-bonded pair has to have at least as many selected atoms mdl1.env.edat.nonbonded_sel_atoms=1 sched = autosched.loop.make_for_model(mdl1) #only optimize the selected residue (in first pass, just atoms in selected #residue, in second pass, include nonbonded neighboring atoms) #set up the mutate residue selection segment s = Selection(mdl1.chains[chain].residues[respos]) mdl1.restraints.unpick_all() mdl1.restraints.pick(s) s.energy() s.randomize_xyz(deviation=4.0) mdl1.env.edat.nonbonded_sel_atoms=2 optimize(s, sched) #feels environment (energy computed on pairs that have at least one member #in the selected) mdl1.env.edat.nonbonded_sel_atoms=1 optimize(s, sched) s.energy() #give a proper name mdl1.write(file=modelname+restyp+respos+'.pdb') #delete the temporary file os.remove(modelname+restyp+respos+'.tmp') -
Then, to introduce the R1275Q mutation into the model, use the command terminal to (1) cd to the directory of the tutorial and (2) run the mutate_model script as shown below:
cd Desktop/modeller-tutorial python mutate_model.py alk_renumbered 1275 GLN A > r1275q.log
We are finally done! The structure should have saved as alk_renumberedGLN1275.pdb. Now you are ready to run an MD simulation so that you can provide the best treatment for your patient!
References
The following are the resources used in creating this tutorial:
- Webb, Benjamin, et al. “Comparative Protein Structure Modeling Using Modeller.”Current Protocols in Bioinformatics (2016).
- Bresler, Scott, et al. “ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma.” Cancer Cell (2014).